专门做母婴的网站有哪些/搜狗推广效果好吗
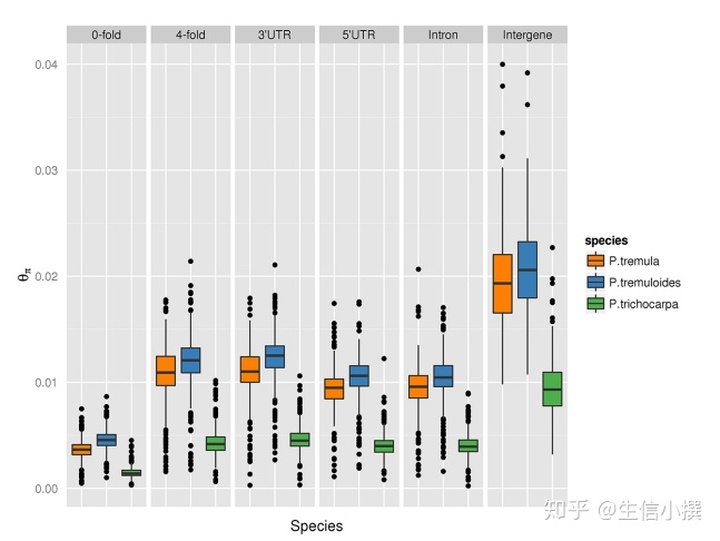
一般我们在进行群体重测序分析的时候,会对vcf文件进行注释,并提取出不同的注释成分,看看他们的核酸多态性(θpi),Tajima’s D之类的参数进行计算,看看在编码区跟非编码区这些参数有什么区别。
本文测试文件请自行下载snpEff,里面的example就可以进行测试
本次小课堂内容就是教会大家最简单的处理方法:首先我们需要准备的内容有几个,第一个是注释软件snpEff
https://sourceforge.net/projects/snpeff/files/snpEff_latest_core.zip/downloadsourceforge.net第二个是参考基因组跟注释文件,模式物种的可以直接下载,自己测的基因组的,就需要自己构建数据库了,构建数据库的方法也非常简单
首先,在下载完的snpEff文件夹里面建立一个文件夹,我愿称之为data(好吧,他只能叫data)
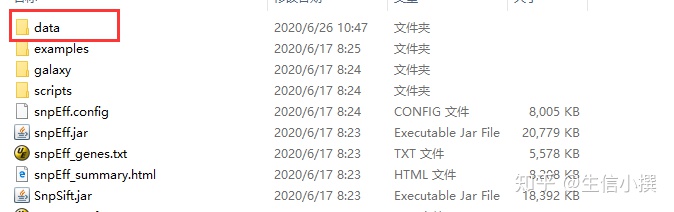
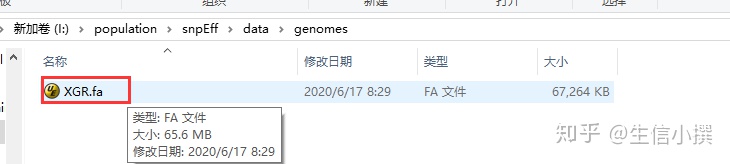
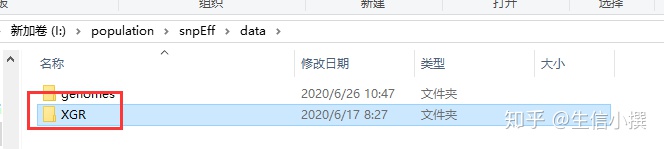
接着,在data文件夹里面创建两个文件夹,一个名字叫genomes,里面放着基因组,然后再创建一个文件夹,文件夹命名为参考基因组名字的前缀,里面放着gtf文件,命名为genes.gtf或者是gff尾缀
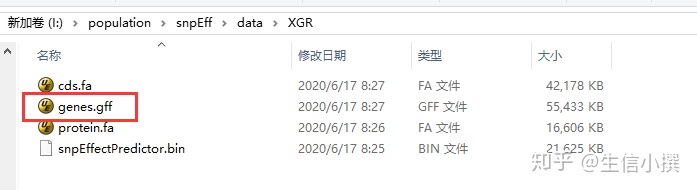
接着回去修改配置文件
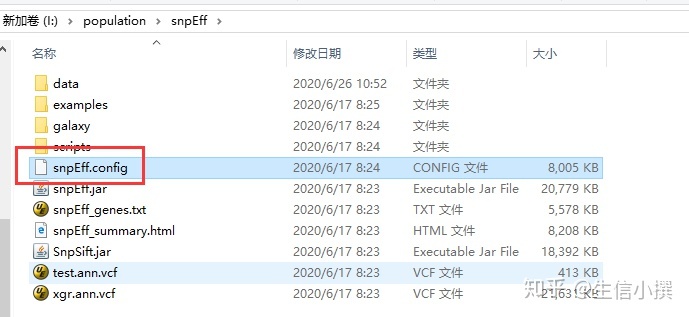
将第三方的信息写进配置文件
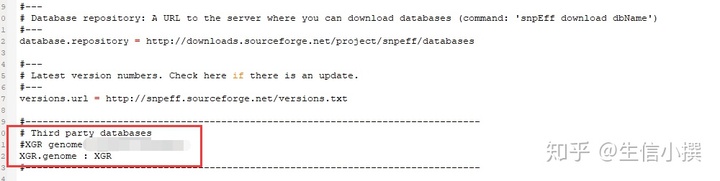
配置完成之后,在命令下建立数据库
java -jar snpEff.jar build -gff3 -v XGR建立完成之后,在gtf文件夹中会出现一个bin
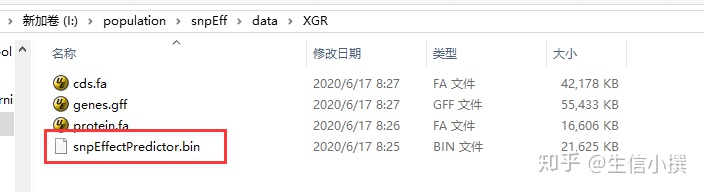
这样我们就可以对vcf文件进行注释了
java -Xmx64g -jar ../snpEff/snpEff.jar eff -v XGR -i vcf test.recode.vcf > test.ann.vcf运行完成之后,就得到了注释文件,可以打开来看看
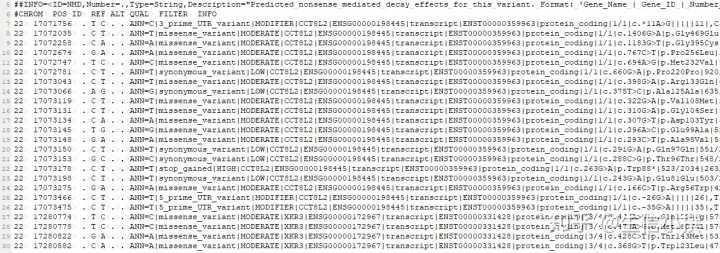
接着就是分离不同的成分,最简单的,如果我想分离3'UTR,5'UTR,intron,intergenic还有synonymous,这些东西,我要怎么搞
其实用python的话,速度勉强能接受,几分钟就过滤完了,代码很简单,跟着我一起完成它
import sysfile = sys.argv[1] #读入命令行参数的第二个元素raw_ann_vcf = open(file, "r") #读入vcf文件lines = raw_ann_vcf.readlines() #逐行读取for line in lines:with open("intergenic_region.vcf","a+") as inter_output:if str("#") in line: #写入头文件inter_output.write(line)if str("intergenic_region") in line: #这里需要自行查阅一下各种注释在vcf的注释字符inter_output.write(line)
raw_ann_vcf.close()需要提取什么内容,直接修改就行,也可以一次性写多个,基本思路就是根据字符串提取
提取完成之后,我们以计算pi为例子讲解下去
使用到的工具是vcftools,命令行很简单
vcftools --vcf intergenic.vcf --keep group1.list --window-pi 30000 --out group1.int
vcftools --vcf intergenic.vcf --keep group2.list --window-pi 30000 --out group2.int
vcftools --vcf intergenic.vcf --keep group3.list --window-pi 30000 --out group3.int
#以30kb的窗口进行计算最后导入R里面绘图
#读入数据
p1 <- read.table("group1.int.windowed.pi", header = T)
p2 <- read.table("group2.int.windowed.pi", header = T)
p3 <- read.table("group3.int.windowed.pi", header = T)
#按照群体命名
p1$pop <- "group1"
p2$pop <- "group2"
p3$pop <- "group3"#合并三个文件
df <- rbind(p1,p2,p3)#画图
library(ggplot2)
options(scipen = 200)
ggplot(df, aes(x=pop, y = PI, fill = pop)) +geom_boxplot() +theme_classic()
如果是展示多个,可以使用grid或者facet_wrap进行展示,效果如下